|
|
Developmental biology - Gene function
New target in Progeria Syndrome
Cambridge University identifies potential new target for devastating genetic disease Hutchinson-Gilford Progeria Syndrome...
Mouse studies by scientists from the University of Cambridge, United Kingdom, have identified a potential therapy target in the devastating genetic disease Hutchinson-Gilford Progeria Syndrome (HGPS), characterised by premature ageing.
In a paper published in Nature Communications, preclinical data shows genetic deregulation of the enzyme N-acetyltransferase 10 (NAT10) leads to significant health and lifespan gains in a mouse model of HGPS.
HGPS is a rare condition with patients having an average life expectancy of 15 years, suffering with symptoms including short stature, low body weight, hair loss, skin thickening, problems with fat storage, osteoporosis, and cardiovascular disease. Patients typically die from a heart attack.
HGPS arises from a specific mutation in the gene encoding a protein called Lamin A.
Cells perceive and relay outside mechanical forces onto the nucleus of the cell through the envelope surrounding the nucleus.
The effect of lowering the nuclear envelope's stiffness impacts mechanical forces and relationships between chromosomes. A softer envelope makes for significant transcription imbalances. Nuclear lamins that regulate chromosome positions, become mislocated in the nuclear interior Nucleic Acids Research.
This leads to production of a shorter and dysfunctional Lamin A protein, specifically in the membranes surrounding the cell nucleus. Disorganization in the chromatin surrounding our DNA, changes how DNA is translated into RNA. Continued accumulate of damage as a cell proliferates affects growth in the child.
By screening molecules in vitro for an effect on nuclear membranes in human HGPS patient-derived cells, the authors had identified a small molecule called remodelin as a potentially effective agent. They then identified which component of the cell was being affected by remodelin - an enzyme called NAT10 with a variety of cell functions.
The aim in the new study was to apply these findings to a mouse model with the same genetic defect as HGPS patients, and see if inhibiting NAT10 - either chemically by administration of remodelin or genetically by engineering reduced production of NAT10 - would
improve the disease.
Results did indeed significantly improve the health of the diseased mice. Diseased mice had increased lifespan with reduced effects of the HGPS mutation across a variety of measures in their tissues as well as at their cellular level.
The research was led by Dr Gabriel Balmus from the Wellcome Trust/ Cancer Research UK Gurdon Institute and Dr Delphine Larrieu from the Cambridge Institute for Medical Research, University of Cambridge; and Dr David Adams from the Wellcome Sanger Institute.
Senior author Professor Steve Jackson: "We're very excited by the possibility that drugs targeting NAT10 may, in future, be tested on people suffering from HGPS. I like to describe this approach as a 're-balancing towards a healthy state'. We first studied the cell biology to understand how the disease affects cells, and then used those findings to identify ways to re-balance the defect at the whole-organism level. Our findings in mice suggest a therapeutic approach to HGPS and other premature ageing diseases."
Abstract
Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare, but devastating genetic disease characterized by segmental premature aging, with cardiovascular disease being the main cause of death. Cells from HGPS patients accumulate progerin, a permanently farnesylated, toxic form of Lamin A, disrupting the nuclear shape and chromatin organization, leading to DNA-damage accumulation and senescence. Therapeutic approaches targeting farnesylation or aiming to reduce progerin levels have provided only partial health improvements. Recently, we identified Remodelin, a small-molecule agent that leads to amelioration of HGPS cellular defects through inhibition of the enzyme N-acetyltransferase 10 (NAT10). Here, we show the preclinical data demonstrating that targeting NAT10 in vivo, either via chemical inhibition or genetic depletion, significantly enhances the healthspan in a LmnaG609G HGPS mouse model. Collectively, the data provided here highlights NAT10 as a potential therapeutic target for HGPS.
Authors: Gabriel Balmus, Delphine Larrieu, Ana C. Barros, Casey Collins, Monica Abrudan, Mukerrem Demir, Nicola J. Geisler, Christopher J. Lelliott, Jacqueline K. White, Natasha A. Karp, James Atkinson, Andrea Kirton, Matt Jacobsen, Dean Clift, Raphael Rodriguez, Sanger Mouse Genetics Project, David J. Adams & Stephen P. Jackson
This study was funded by the Wellcome and the Medical Research Council, and core funding to the Gurdon Institute from the Wellcome and Cancer Research UK.
Return to top of page
| |
|
Apr 30, 2018 Fetal Timeline Maternal Timeline News News Archive
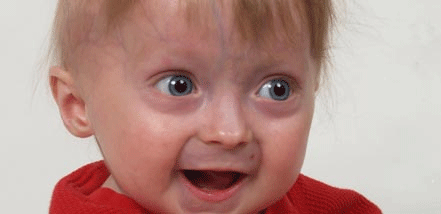 Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare, devastating genetic disease characterized by premature aging. Cells from patients accumulate progerin, a toxic form of Lamin A, disrupting the shape and chromatin organization of a cell nucleus, leading to accumulated DNA-damage and death. Image credit: Progeria Research Foundation.
|


