|
|
Developmental Biology - Heart Cell Regeneration
Heart Mitochondria Fuel Regeneration
New study suggests encouraging cardiomyocytes to digest glucose, instead of fatty acids, could improve treatment of heart failure...
Switching what the powerhouses of heart cells consume for energy could help the heart regenerate when cells die, a new study led by UT Southwestern researchers suggests. The finding, published in the Feb. 20, 2020, Nature Metabolism, could open whole new avenues for treating a variety of conditions in which heart muscle becomes damaged, including heart failure caused by viruses, toxins, high blood pressure, or heart attacks.
Current pharmaceutical treatments for heart failure - including ACE inhibitors and beta blockers - center on trying to stop a vicious cycle of heart muscle loss as strain further damages remaining heart muscle, causing more cells to die, explains University of Texas (UT) Southwestern physician-researcher Hesham A. Sadek MD PhD, the J. Fred Schoellkopf, Jr. Chair in Cardiology. There are no existing treatments for rebuilding heart muscle.
Nine years ago, Sadek and his colleagues discovered that mammalian hearts can regenerate if damaged in the first few days of life, spurred by the division of cardiomyocytes, cells responsible for a heart's contractile ability. However, this capacity is completely lost by 7 days old, an abrupt turning point in which division of these cells dramatically slows.
Subsequent research has shown that this change in regenerative capacity appears to stem, at least in part, from damaging free radicals generated by organelles known as mitochondria, which power cells. These free radicals damage cells' DNA, a phenomenon called DNA damage, which prompts them to stop dividing.
The shift in free radical production appears to occur when mitochondria cardiomyocytes change what they consume for energy.
In utero mitochondria rely on glucose, however at birth they begin the switch to fatty acids with their adaptation to energy-dense molecules found in breast milk.
Sadek and colleagues wondered whether forcing mitochondria to continue to consume glucose might impede DNA damage and possibly extend the window for heart cell regeneration. To test this idea, researchers tried the following experiments:
First Experiment
Researchers followed mouse pups whose mothers were genetically altered to produce low-fat breastmilk and that fed on low-fat chow after they weaned. Researchers found that these rodents' hearts maintained regenerative capacity weeks later than normal. Their cardiomyocytes continued to express genes associated with cell division for significantly longer than mice fed a diet of regular breastmilk and chow.
However, the effect didn't last into adulthood as their livers eventually synthesized fats their diets were missing, making up the deficit significantly reducing their hearts' regenerative capacity.
Second Experiment
Researchers then genetically deleted the enzyme in mice known as pyruvate dehydrogenase kinase 4 (PDK4), which heart cellmitochondria need to digest fatty acids. When researchers turned off PDK4 production, mouse cardiomyocytes adapted by consuming glucose instead of fatty acids even into adulthood.
After simulating a heart attack in these mice, it was found the animals' heart function improved! This was accompanied by a gene expression marker suggesting their cardiomyocytes were still actively dividing.
Sadek points out these findings are proof of principle, to reopen the question of heart cell regeneration through manipulation of energy consumption by cardiomyocyte mitochondria.
"Eventually it may be possible to develop drugs that change what cardiomyocytes eat to make them divide again, reversing heart failure and representing a true cure."
Hesham A. Sadek MD PhD, UT Southwestern, Dallas, Texas, USA; physician-researcher and J. Fred Schoellkopf, Jr. Chair in Cardiology.
Abstract
The neonatal mammalian heart is capable of regeneration for a brief window of time after birth. However, this regenerative capacity is lost within the first week of life, which coincides with a postnatal shift from anaerobic glycolysis to mitochondrial oxidative phosphorylation, particularly towards fatty-acid utilization. Despite the energy advantage of fatty-acid beta-oxidation, cardiac mitochondria produce elevated rates of reactive oxygen species when utilizing fatty acids, which is thought to play a role in cardiomyocyte cell-cycle arrest through induction of DNA damage and activation of DNA-damage response (DDR) pathway. Here we show that inhibiting fatty-acid utilization promotes cardiomyocyte proliferation in the postnatal heart. First, neonatal mice fed fatty-acid-deficient milk showed prolongation of the postnatal cardiomyocyte proliferative window; however, cell-cycle arrest eventually ensued. Next, we generated a tamoxifen-inducible cardiomyocyte-specific pyruvate dehydrogenase kinase 4 (PDK4) knockout mouse model to selectively enhance oxidation of glycolytically derived pyruvate in cardiomyocytes. Conditional PDK4 deletion resulted in an increase in pyruvate dehydrogenase activity and consequently an increase in glucose relative to fatty-acid oxidation. Loss of PDK4 also resulted in decreased cardiomyocyte size, decreased DNA damage and expression of DDR markers and an increase in cardiomyocyte proliferation. Following myocardial infarction, inducible deletion of PDK4 improved left ventricular function and decreased remodelling. Collectively, inhibition of fatty-acid utilization in cardiomyocytes promotes proliferation, and may be a viable target for cardiac regenerative therapies.
Authors
Alisson C. Cardoso, Nicholas T. Lam, Jainy J. Savla, Yuji Nakada, Ana Helena M. Pereira, Abdallah Elnwasany, Ivan Menendez-Montes, Emily L. Ensley, Ursa Bezan Petric, Gaurav Sharma, A. Dean Sherry, Craig R. Malloy, Chalermchai Khemtong, Michael T. Kinter, Wilson Lek Wen Tan, Chukwuemeka G. Anene-Nzelu, Roger Sik-Yin Foo, Ngoc Uyen Nhi Nguyen, Shujuan Li, Mahmoud Salama Ahmed, Waleed M. Elhelaly, Salim Abdisalaam, Aroumougame Asaithamby, Chao Xing, Mohammed Kanchwala, Gonçalo Vale, Kaitlyn M. Eckert, Matthew A. Mitsche, Jeffrey G. McDonald, Joseph A. Hill, Linzhang Huang, Philip W. Shaul, Luke I. Szweda and Hesham A. Sadek.
Acknowledgements
H. I. May (Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA) for the myocardial infarction surgeries in mice. H.A.S. is supported by grants from the NIH (1R01HL115275 and 5R01H2131778), National Aeronautics and Space Administration (NNX-15AE06G), American Heart Association (16EIA27740034), Cancer Prevention and Research Institute of Texas (RP160520), Hamon Center for Regenerative Science and Medicine and Fondation Leducq. N.T.L. is supported by a Haberecht Wildhare-Idea Research Grant. A.D.S. is supported by grant from the NIH (R37-HL034557), C.R.M. is supported by grant from the NIH (P41-EB015908) and G.S. is supported by the grant from the AHA (18POST34050049). M.K. is supported by the grants from NIH (3P20GM103447 and 5P30AG050911). I.M.M. is supported by Alfonso Martin Escudero Foundation Fellowship. N.U.N.N. is supported by AHA Postdoctoral Fellowship 19POST34450039. J.A.H. is supported by grants from the NIH (HL-120732, HL-128215 and HL-126012).
Return to top of page.
| |
|
Mar 31 2020 Fetal Timeline Maternal Timeline News
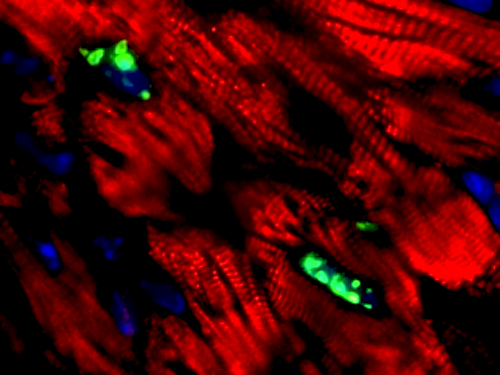 pH3 positive cardiomyocytes. CREDIT Hesham A. Sadek MD PhD, UT Southwestern.
|


