|
|
Developmental Biology - Methylation
Methylation Shapes Our Genome
Mammal embryos need precise matching of methyl groups to genes in order to regulate gene function. Mistakes in methyl attachments, are now commonly associated with gene mutations and diseases resulting from these errors...
Salk researchers have now generated 168 new maps of chemical marks on strands of DNA taken from mice. These new maps are of chemical tags called 'methylation marks'. Where they occur on DNA strands, pinpoints gene regions corresponding to developmental disorders. The new methylation maps follow changes in mouse embryos in utero to birth, giving science and medicine a more precise picture of when - and where - developmental disorders occur.
This new data just published July 29, 2020, in a special edition of Nature is devoted to the ENCODE Project. ENCODE is a public research effort aimed at identifying all functional elements in the human and mouse genomes.
Creating these intricate maps can help narrow down regions on the human genome that play a role in diseases such as schizophrenia and Rett Syndrome. The paper's authors are also authors on two additional papers in this special edition of Nature.
"This is the only available data set that looks at the methylation in a developing mouse over time, tissue by tissue. It's going to be a valuable resource to help narrow down the causal tissues of human developmental diseases."
Joseph Ecker PhD, senior author, professor and Howard Hughes Medical Institute Investigator; Salk Genomic Analysis Laboratory, Salk Institute for Biological Studies, La Jolla, California, USA.
While the sequence of DNA contained in every cell of your body is virtually identical, chemical marks on those strands of DNA give cells their unique identities. The patterns of methylation on adult brain cells, for instance, are different than those on adult liver cells. This is in part because short stretches in the genome called 'enhancers'.
When transcription factor proteins bind to these enhancer regions, a target gene is much more likely to be expressed or made functional. However, when an enhancer is methylated, transcription factors generally can't bind and the associated gene is less likely to become activated.
Methyl marks are akin to applying the hand brake after parking a car.
Researchers know that mutations in enhancer regions by affecting the expression [function] level of a corresponding gene can cause disease. But, there are hundreds of thousands of enhancers and they can be located far from the gene they help regulate. So narrowing down which enhancer mutations might play a role in a developmental disease has been a challenge.
In the new work, Ecker and collaborators used experimental technologies and computational algorithms they had previously developed to study DNA methylation patterns in cell samples over a dozen types of tissues from mice and over eight embryo developmental stages.
"The breadth of samples that we applied this technology to is what is really key."
Yupeng He PhD, first author and previously a Salk postdoctoral research fellow, now a senior bioinformatics scientist at Guardant Health, Inc.
They discovered more than 1.8 million regions of the mouse genome had variations in methylation based on tissue, developmental stage or both.
Early in development, those changes were mostly the loss of methylation on DNA akin to removing the hand brake on gene expression and allowing developmental genes to turn on and become active.
After birth, however, most sites became highly methylated again, putting the brakes back on gene expression, as the mouse approaches birth.
"We think that removal of methylation marks, makes the whole genome more open to dynamic regulation during development. After birth, genes critical for early development need to be stably silenced as we don't want them turned on in mature tissue. That is when methylation steps in to help shut down enhancers of early development."
Yupeng He PhD,
Genomic Analysis Laboratory, The Salk Institute for Biological Studies; and Bioinformatics and Systems Biology Program, University of California, San Diego, La Jolla, California, USA.
In the past, many researchers studied methylation by homing in on areas of the genome near genes called CpG islands sections of DNA with a lot of cytosine and guanine base pairs. Typical methylation occurs when a methyl is added to a cytosine that's followed by a guanine. However, the new work found in development 91.5 % of methylation sites are far from CpG islands.
"If you only look at those CpG island regions near genes, as many people do, you'll miss a lot of the meaningful DNA changes that could be directly related to your research questions."
Yupeng He PhD
Highlights
CMS121, a fisetin-derivative, alleviates memory decline in a double transgenic AD mouse model.
CMS121 is able to reduce lipid peroxidation and neuroinflammation, both in vitro and in vivo.
We identify fatty acid synthase (FASN), which shows increased protein levels in human AD patients, as a target of CMS121.
Our results confirm the involvement of lipid peroxidation and perturbed lipid metabolism in AD pathophysiology.
Decreasing lipid levels through FASN inhibition can be effective against excess lipid peroxidation.
Abstract
Cytosine DNA methylation is essential for mammalian development but understanding of its spatiotemporal distribution in the developing embryo remains limited1,2. Here, as part of the mouse Encyclopedia of DNA Elements (ENCODE) project, we profiled 168 methylomes from 12 mouse tissues or organs at 9 developmental stages from embryogenesis to adulthood. We identified 1,808,810 genomic regions that showed variations in CG methylation by comparing the methylomes of different tissues or organs from different developmental stages. These DNA elements predominantly lose CG methylation during fetal development, whereas the trend is reversed after birth. During late stages of fetal development, non-CG methylation accumulated within the bodies of key developmental transcription factor genes, coinciding with their transcriptional repression. Integration of genome-wide DNA methylation, histone modification and chromatin accessibility data enabled us to predict 461,141 putative developmental tissue-specific enhancers, the human orthologues of which were enriched for disease-associated genetic variants. These spatiotemporal epigenome maps provide a resource for studies of gene regulation during tissue or organ progression, and a starting point for investigating regulatory elements that are involved in human developmental disorders.
Authors
Yupeng He, Manoj Hariharan, David U. Gorkin, Diane E. Dickel, Chongyuan Luo, Rosa G. Castanon, Joseph R. Nery, Ah Young Lee, Yuan Zhao, Hui Huang, Brian A. Williams, Diane Trout, Henry Amrhein, Rongxin Fang, Huaming Chen, Bin Li, Axel Visel, Len A. Pennacchio, Bing Ren and Joseph R. Ecker.
Acknowledgements
The authors thank J. Li, S. C. Huang, E. A. Mukamel and L. Song for critical comments. D.U.G. is supported by the A.P. Giannini Foundation and NIH IRACDA K12 GM068524. A.V. and L.A.P. were supported by National Institutes of Health grant U54HG006997, and the research conducted at the E. O. Lawrence Berkeley National Laboratory was performed under Department of Energy Contract DE-AC02-05CH11231, University of California. J.R.E. is an Investigator of the Howard Hughes Medical Institute. Use of the Extreme Science and Engineering Discovery Environment (XSEDE) was supported by National Science Foundation grant number ACI-1548562.
This work was supported by the National Institutes of Health ENCODE Project (U54 HG006997). The data that support these findings are publicly accessible at Nature: Spatiotemporal DNA methylome dynamics of the developing mouse fetus and
Spatiotemporal DNA Methylome Dynamicsof the Developing Mammalian Fetus
from the Salk Institutes. Additional RNA-seq data sets are available at the NCBI Gene Expression Omnibus (accession GSE100685). Further details describing the data used in this study can be found in Supplementary Tables 1 and 2.
About the Salk Institute for Biological Studies:
Every cure has a starting point. The Salk Institute embodies Jonas Salk's mission to dare to make dreams into reality. Its internationally renowned and award-winning scientists explore the very foundations of life, seeking new understandings in neuroscience, genetics, immunology, plant biology and more. The Institute is an independent nonprofit organization and architectural landmark: small by choice, intimate by nature and fearless in the face of any challenge. Be it cancer or Alzheimer's, aging or diabetes, Salk is where cures begin. Learn more at: salk.edu.
The work and the researchers involved were supported by grants from the A.P. Giannini Foundation, the National Institutes of Health, the Department of Energy, the Howard Hughes Medical Institute, and the National Science Foundation.
The other two Nature special edition papers are Moore et al., "Expanded Encyclopedias of DNA Elements in the Human and Mouse Genomes" and Gorkin et al., "An Atlas of Dynamic Chromatin Landscapes in Mouse Fetal Development."
The authors declare they have no competing interests.
Return to top of page.
|
|
Aug 11 2020 Fetal Timeline Maternal Timeline News
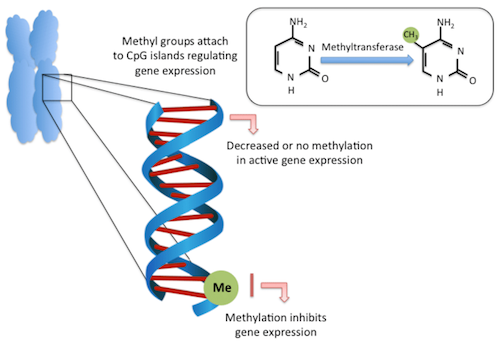 DNA MethylationThe methylation of cytosine. Methylation occurs mainly on the 5th carbon of the cytosine base, forming 5-methylcytosine (5-mC). Mainly occurring around CpG clusters (CpG islands) at gene promoter regions associated with gene inactivation. CREDIT Research Gate
|
|


